Chondrosarcoma is a family of cartilage-producing bone cancers and the third most common primary bone sarcoma. Most chondrosarcomas grow slowly, metastasise in only 15–25% of cases and are primarily treated by surgery with the goal of clear margins and lasting cure.
30–60 yrs
Typical age range
83%
10-yr survival, Grade I (ACT)
Pelvis
Most common site
What is chondrosarcoma?
Chondrosarcomas arise from cartilage cells in bone. They typically present as a slow-growing, painful mass and behave very differently from the other bone sarcomas — most do not respond to chemotherapy or radiotherapy, making complete surgical removal the key to cure.
Key point:A slow-growing cancer that mainly requires surgical treatment for cure.
Who gets chondrosarcoma?
Age: Most common between 30 and 60 years.
Gender: Slight male predominance.
Common sites: Pelvis (most common), long bone shaft & metaphysis, shoulder girdle and — rarely — the spine.
Origin: May arise de novo (primary) or from a pre-existing enchondroma / osteochondroma (secondary).
Red flags
When to seek specialist review
Dull, persistent bone pain
Aching that lasts months — often the only early symptom.
Rapidly growing mass
A previously stable lump that suddenly starts enlarging.
New pain in a known benign tumour
Pain developing in an enchondroma or osteochondroma needs review.
Pelvic / shoulder girdle ache
Deep, vague pain in the pelvis or shoulder that does not settle.
Types of chondrosarcoma
The chondrosarcoma family includes several subtypes that look, behave and respond to treatment differently. Subtype is decided by an experienced bone-tumour pathologist.
Conventional (85%)
The commonest form — central (within bone), peripheral (from bone surface) or juxtacortical.
Dedifferentiated (10%)
High-grade and aggressive — a low-grade cartilage tumour with an abrupt high-grade non-cartilaginous component.
Mesenchymal (<2%)
High-grade variant of young adults — chemotherapy-responsive with up to 89% 10-year survival in multimodal therapy.
Clear cell (<2%)
Low-grade — typically affects the ends of long bones (epiphysis).
Grading
Why grade matters
Tumour grade is the single most important factor determining treatment and survival.
Atypical Cartilaginous Tumour
Grade I (ACT)
83%
10-year survival
Rarely metastasises (<5%). Often managed with extended curettage.
Intermediate-grade
Grade II
64%
10-year survival
Wide surgical excision with clear margins.
High-grade
Grade III
30%
10-year survival
Aggressive — wide excision and multidisciplinary planning.
Grade IV-equivalent
Dedifferentiated
28%
10-year survival
Most aggressive — chemotherapy of limited benefit.
Diagnosis
How is chondrosarcoma diagnosed?
Imaging followed by a planned biopsy at a specialist centre. An improperly placed biopsy can compromise future limb salvage.
X-rays
Show characteristic cartilage 'rings and arcs' calcification with endosteal scalloping or cortical destruction.
MRI / CT
Defines tumour extent within bone and soft tissue — essential for surgical planning.
PET-CT
Assesses metabolic activity and detects metastases. Can occasionally avoid biopsy and guide treatment.
Core needle biopsy
Confirms diagnosis — sometimes PET-CT guided to sample the most active area for accurate grading.
Clinical clue: dull, aching pain over 3–6 months with a palpable, tender mass — especially in the pelvis or shoulder girdle — warrants urgent imaging.
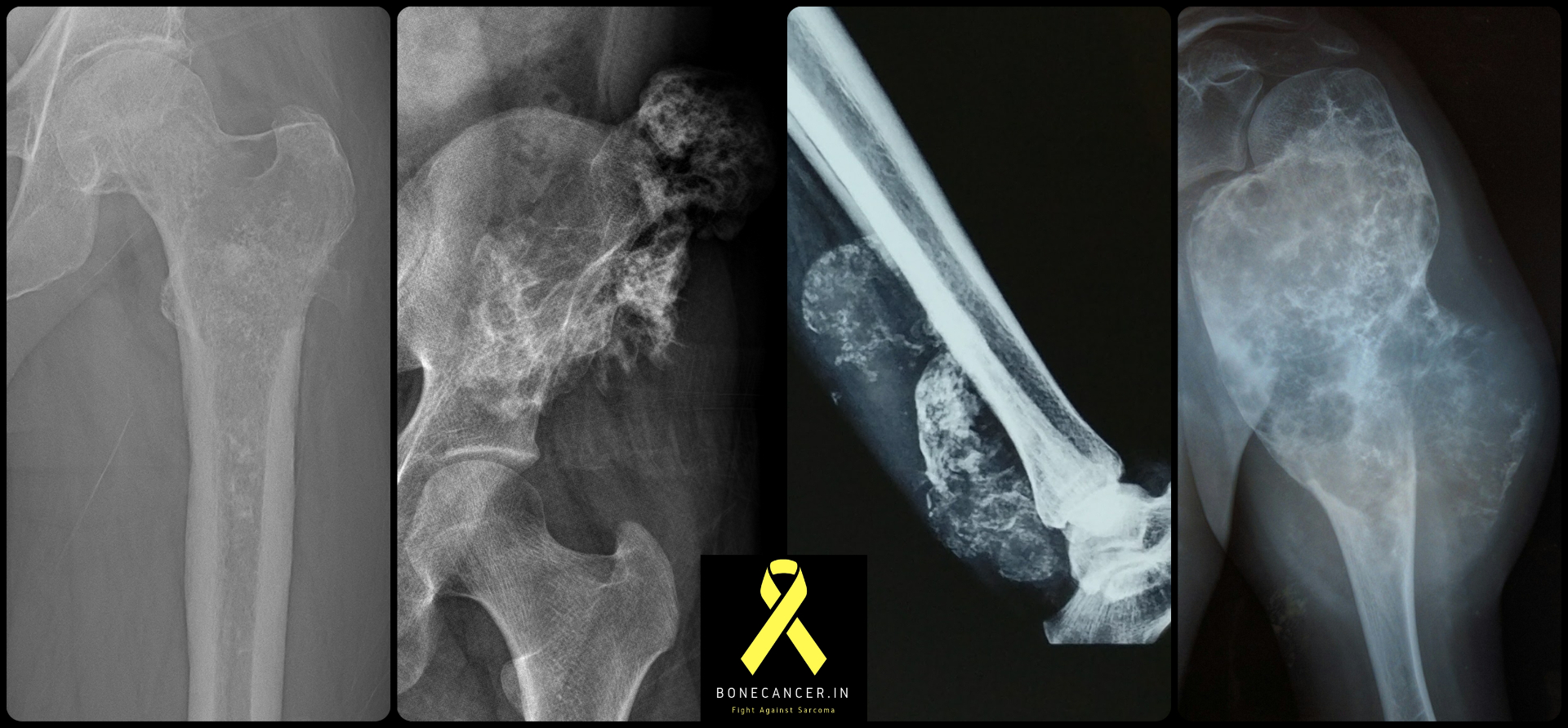
Representative radiographs of chondrosarcoma across common sites — proximal femur, pelvis, distal forearm and proximal humerus. Note the characteristic ring-and-arc cartilage mineralisation, endosteal scalloping, cortical breach and soft-tissue mass.
Treatment
Surgery is the primary treatment
The approach is tailored to grade, location and subtype. Most cures are achieved by careful surgery with adequate margins.
Low-grade in limbs
Extended curettage
For Grade I / Atypical Cartilaginous Tumours of the appendicular skeleton.
High-speed burr to extend the curettage margin
Optional chemical adjuvants (phenol, liquid nitrogen)
Cavity filled with bone graft or cement
Low local recurrence with this approach
Higher grade / axial
Wide en-bloc excision
For pelvic, spinal and higher-grade tumours — the primary curative procedure.
Tumour prosthesis (artificial joint or bone)
Biological reconstruction (donor or recycled bone)
Graft–prosthetic composite combinations
Soft-tissue reconstruction or planned pseudoarthrosis where appropriate
Chemotherapy
Selective use only
Conventional chondrosarcoma is generally resistant to chemotherapy.
Mesenchymal subtype: responds well to anthracycline + ifosfamide
Dedifferentiated subtype: limited benefit, mainly palliative role
Used in multidisciplinary protocols at specialist centres only
Radiotherapy
Limited but useful role
Slow-growing tumours respond poorly, but radiotherapy is valuable in selected scenarios.
Unresectable tumours or positive surgical margins
Doses often need to exceed 60 Gy
Proton/heavy-ion therapy considered for axial sites
Real cases · Surgical management
Pelvic & hip-bone chondrosarcoma — case videos
Limb- and function-preserving surgery for chondrosarcoma in some of the most challenging anatomical regions.
Pelvic chondrosarcoma
Chondrosarcoma of the pelvis — surgical management
Approach to a chondrosarcoma involving the pelvis — wide resection and reconstruction planning in a complex anatomical region.
Hip-bone chondrosarcoma
Chondrosarcoma of the hip bone — reconstruction
Limb- and joint-preserving surgery for chondrosarcoma involving the hip bone with reconstructive options to restore function.
Pelvic — no reconstruction
Chondrosarcoma of pelvic bone without reconstruction
Wide excision of a pelvic chondrosarcoma where planned pseudoarthrosis (no bony reconstruction) provides excellent functional outcome.
Real-life patient examples
Case 1 · Grade I chondrosarcoma
Hanumanthaiah, 45 — humerus
A 45-year-old teacher noticed arm pain for 6 months. X-rays showed a cartilage tumour in the upper arm bone; biopsy confirmed Grade I chondrosarcoma. He underwent extended curettage with cement filling and returned to full activities within 3 months. Five years later he remains cancer-free with excellent arm function.
Case 2 · Dedifferentiated chondrosarcoma
Pushpa, 55 — pelvis
A 55-year-old homemaker developed severe hip pain and a rapidly growing mass. MRI revealed a large pelvic tumour with soft tissue extension; biopsy confirmed dedifferentiated chondrosarcoma. She underwent extensive pelvic resection with reconstruction followed by chemotherapy. Despite the aggressive disease, multimodal care provided meaningful disease control and ongoing surveillance.
If chondrosarcoma comes back locally
High-grade tumours require wide re-excision
Grade I tumours can often be retreated with repeat curettage